Respiratory Ward - Histiocytosis X

Cases from the Respiratory Ward
DISCUSSION: PULMONARY LANGERHANS CELL HISTIOCYTOSIS - CASE PRESENTATION
Mr AL
35 year old male resident of Soweto.Was previously well and now presents with a 6 week history of a dry cough associated with decreased effort tolerance.No history of weight loss or night sweats. He is a smoker of 20 cigarettes per day for the past 15 yrs. He works in a factory that makes foam rubber mattresses. Otherwise he has been well.
On Examination, the patient was not distressed. He was hemodynamically stable. His respiratory rate was 30. He was not cyanosed nor was he clubbed. On cardiac examination there were no signs of pulmonary hypertension. On respiratory examination, the breath sounds were normal and there were a few, scattered end inspiratory crackles bilaterally. The rest of his examination was normal.
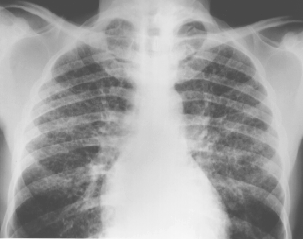
Relavent investigations were as follows;( CXR and CT scan are below )
- pO2 63 mmHg
- pCO2 31 mmHg
- Full blood count, electrolytes and liver functions were normal
The Pulmonary function tests showed evidence of obstruction;
- FVC: 2.39 (71%)
- FEV1: 1.33 (47%)
- FEV1/FVC: 56%
- TLC: 5.14 (105%)
- RV: 2.80 (234%)
- DLCO: 16.26 (78%)
- DLVA: 3.97 (93%)
The patient was not subjected to an open lung biopsy as he absconded from hospital. However, a presumptive diagnosis of histiocytosis X was made on the basis of the CXR and CT Scan of the chest aswell as the clinical picture.
CASE DISCUSSION
Definition: the histiocytoses are an ill understood and heterogeneous group of syndromes, characterised by an abnormal proliferation of histiocytes. A classification of the histiocytoses was proposed by the Histiocyte Society in 1987, in an attempt to standardize nomenclature.
| HISTIOCYTE SOCIETY CLASSIFICATION | |
| CLASS 1 |
|
| CLASS 2 |
|
| CLASS 3 |
|
| CLASS 4 |
|
This discussion will deal with pulmonary Langerhans Cell Histiocytosis (LCH). Previously LCH was called Histiocytosis X, and was classified according to;
- GENERALISED DIFFUSE ( LETTERER-SIWE)
- GENERALISED MULTIFOCAL (HAND-SCHULLER-CHRISTIAN)
- LOCALISED (EOSINOPHILIC GRANULOMA)
This classification and the syndromes mentioned in it are now considered to be outmoded, and should no longer be used.
In spite of the wide clinical spectrum of disease in LCH, the pathological lesions are uniform in all forms. Further discussion will be centered on pulmonary LCH.
EPIDEMIOLOGY
Pulmonary LCH is a rare condition. In a series of 502 open lung biopsies for chronic , diffuse , infiltrative lung disease ? LCH was identified in 3.4%. Isolated pulmonary disease affects predominantly young and middle aged adults. Over 90% of patients are smokers and approximately twice as many males as females are affected.
PATHOGENESIS AND AETIOLOGY
The pathogenesis has not been fully elucidated, but numerous theories have been proposed.
The disease process appears to fit well with and uncontrolled immune response to an unknown antigen, initiated or propagated by Langerhans cells (LC).
2 classes of antigen presenting cells are found in the lung: monocyte/macrophage and dendritic cell/ langerhans cell lineages. The LC are extremely potent antigen presenting cells which express peptide antigen in association with their own HLA molecules for recognition by T cells as well as secreting IL-1 and PGE-2.
LC are derived from dendritic cells and form a continuous network intercalated between epithelial cells in intraparenchymal airways. They are identified by the expression of CD 1a surface antigen and by the presence of pentalaminar Birbeck granules, which look like tennis raquets on electron microscopy.
Cigarette smoking can double the number of LC in the parenchymal airways ( but not in larger airways), probably by increasing their recruitment to the lung and accelerating differentiation from dendritic cells to LC.
Langerhans cells accumulate preferentially at sites of epithelial hyperplasia, possibly following secretion of the cytokine GM-CSF by the hyperplastic epithelium.
The reasoning that LCH is an abnormal immune response to an antigen is based on the following arguments:
- LC and lymphocytes are the predominant cell types in early lesions- and the only described function of LC is to act as apc.
- The evolution of pathological changes follows the same steps as other granulomatous processes, namely initial predominance of apc and lymphocytes followed by infiltration by inflammatory cells and then progressive replacement by macrophages and fibrosis.
- No other pathogenetic mechanism explains the findings as well
Immediately the question arises as to what the possible antigen could be. The bronchiolar distribution and the high incidence of smoking in affected patients is suggestive of an inhaled antigen. However this does not explain disease in non smokers, and extrapulmonary disease.
A proposed theory is that the bronchial epithelial cells could serve as the antigen. Epithelial hyperplasia is known to be associated with increased numbers of LC at these sites and the hyperplastic epithelium could express neoantigens ( stress proteins) ? not present in normal epithelial cells, that would serve as antigenic stimulus. Once the immune response was triggered a vicious cycle would be triggered with epithelial destruction, and release of immune mediators inducing hyperplasia of adjacent epithelium, leading to further destruction.
This would account for the high incidence of affected smokers as cigarette smoke induces epithelial hyperplasia.
PATHOLOGICAL FINDINGS
Characteristic pathological findings consist of destructive granulomatous lesions, occurring around bronchioles, with normal pulmonary parenchyma lying between granulomas. As previously mentioned granulomas evolve through stages: from initial LC plus lymphocytes to infiltration with inflammatory cells, to end stage lesions with rare LC, and numerous pigment laden macrophages and fibrosis.
Light microscopy and EM studies of open lung biopsies from 62 patients (with normal control specimens) showed the importance of luminal fibrosis and elastic fibre degradation in the process of pulmonary structural remodelling.
The steps of structural remodelling are deemed to occur as follows:
- Epith cell damage and detachment from the basement membrane.
- Migration of LC, inflammatory cells and myofibroblasts into intra alveolar spaces.
- Late stage ? formation of extra cellular components leading to obliteration of alveoli and coalescence of alveolar walls.
- Degradation of elastic fibres in alveolar walls adjacent to granulomatous lesions, giving rise to irregularly dilated alveolar spaces.
Reconstruction of granulomas using serial sections suggests that the lesions arise in the terminal bronchiole or the proximal respiratory bronchiole, and with time extends distally, ultimately reaching alveoli. In the same lesion therefore : proximal fibrosis exists with distal cellularity and relative abundance of LC.
CLINICAL FEATURES
Symptoms:
- 60% cough
- 40% dyspnoea
- 10-25% asymptomatic
- 20% pleuritic chest pain ? most commonly due to spontaneous pneumo
- 15-30% constitutional symptoms
- 5-10% extrapulmonary manifestations (bone, diabetes insipidus)
Examination is often unremarkable with crackles heard in only 10-20% of patients and clubbing being seen rarely. Although classified under interstitial lung disease ? from the pathological lesion it may be more appropriately classified as a bronchiolitis, which would also explain many of the clinical features unusual for ILD.
SPECIAL INVESTIGATIONS
Lung function tests show a characteristically mixed or mild restrictive / obstructive picture with decreased transfer factor. Lab tests are unhelpful: ACE and Eosinophil count are normal.
RADIOLOGICAL FINDINGS
CXR:
- diffuse reticulonodular opacities superimposed on multiple small cysts ar ring shadows 5-10 mm in diameter with predominance in mid- upper zones, and sparing the costophrenic angles.
- Changes are not always symmetrical. Lung volumes are preserved or increased ? in contrast to other fibrosing lung disorders.
HRCT chest:
- thin walled lung cysts (<10mm) , some confluent or with bizarre shapes
- nodules (1-5mm) some centrilobular and peribronchial
- upper lobe predominance
- cavitary nodules
- fine reticular opacities, ground glass opacities.
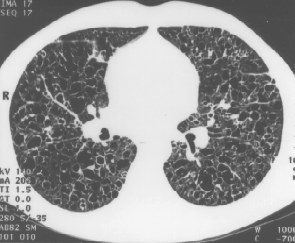
HRCT shows no central / peripheral predominance of lesions. Intervening lung parnchyma appears normal in many patients.
HRCT can differentiate pts with nodules only from other diseases causing pulmonary nodules (sarcoid, silicosis, TB, metastasis), by the centrilobular distribution of nodules and sparing of the costophrenic angles. In addition fibrosis can be distinguished from fibrosis due to CFA by the sparing of the lung bases, the presence of normal surrounding lung parenchyma, and the preservation of lung volumes.
DIAGNOSIS
The diagnosis can often be made with a fair degree of accuracy from the clinical picture, and the radiological features in a young smoker. It is usually confirmed by open lung biopsy ? specimen sent for EM, and occasionally by transbrochiole biopsy. Brochiolar lavage is a less invasive method of making the diagnosis. The presence of > 4% LC among total BAL cells (characterised by positive CD1 immunostaining , or the presence of Birbeck granules on EM) is diagnostic. However BAL has a high false negative rate , being positive in only 50% of sufferers.
PROGNOSIS
Death in PLCH is mainly due to terminal respiratory failure and secondly to the development of pulmonary neoplasms. The natural course is variable: 25% undergo complete remission, 50% stabilise, and 25 % undergo progressive deterioration. Mortality is 2-6%.
A survival analysis was performed on 45 patients with pulmonary LCH. A univariante analysis demonstrated diminished survival associated with:
- older age at diagnosis (26y)
- lower FEV12/FVC ratio (<0,66)
- high RV/TLC ratio (>0,33)
- steroid therapy during follow up
Multivariate analysis showed the FEV1/FVC ratio and age to be of the best prognostic value. Other poor prognostic features include : recurrent pneumothoraces, extensive initial radiological changes, and decreased transfer factor at diagnosis.
Good prognosis was associated with complete cessation of smoking and sparing of the cosphrenic angles. Too few patients underwent HRCT in order for HRCT determinants of survival to be identified.
ASSOCIATED MALIGNANCIES
This is an interesting area with minimal data, but a number of case reports. Malignancy associated with pulmonary LCH appears to occur in a number of different clinical settings, namely:
- Ca lung assoc. with pulmonary LCH ? probably share a common aetiological agent ie smoking
- Discovery of lymphoma with the simultaneous discovery of nodal or extranodal LCH
- Histiocytosis developing following treatment of lymphoma with DXT
- Malignancy following pulmonary LCH possibly representing malignant transformation.
Malignancy is usually Hodgkin?s disease or malignant histiocytosis.
TREATMENT
The only certain therapy is to stop smoking. Because of the high rate of spontaneous remission and rarity of the disease ? no reliable data concerning the efficacy of various treatment schedules on survival exist. To date steroids, chemotherapy(cyclophosphamide,methotrexate,vinblastine,vincristine,chlorambucil) , combination therapy and allogenic bone marrow transplant have been used. Since it is no longer considered to be a malignancy, the use of aggressive chemo is difficult to justify ,in view of the serious side effects.
Current recommendation for patients with single organ disease is a period of close observation with complete cessation of smoking, with consideration for the introduction of steroids or chemotherapy if progression is evident.
REFERENCES
- Cheyne C: Histiocytosis X. J Bone Joint Surg Br 53:366, 1971 87.
- Komp DM, Herson J, Starling KA, et al: A staging system for histiocytosis X: a Southwest Oncology Group study. Cancer 47:798, 1981 88.
- Bunch WG: Orthopedic and rehabilitation aspects of eosinophilic granuloma. Am J Pediatr Hematol Oncol 3:151, 1981 89.
- Dunger DB, Broadben V, Yeoman E, et al: The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 321:1157, 1989 90.
- Greenberger JS, Cassady JR, Jaffe N, et al: Radiation therapy in patients with histiocytosis: management of diabetes insipidus and bone lesions. Int J Radiat Oncol Biol Phys 5:1749, 1979 91.
- Starling KA: Chemotherapy of histiocytosis. Am J Pediatr Hematol Oncol 3:157, 1981 92.
- Ceci A, DeTerlizzi M, Colella R, et al: Etoposide in recurrent childhood Langerhans cell histiocytosis: an Italian cooperative study. Cancer 62:2528, 1988 93.
- Rosai J, Dorfman RF: Sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder: analysis of 34 cases. Cancer 30:1174, 1972 94.
- Foucar E, Rosai J, Dorfman RF: Sinus histiocytosis with massive lymphadenopathy. Arch Otolaryngol 104:687, 1978 95.
- Foucar E, Rosai J, Dorfman R: Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): review of the entity. Semin Diagn Pathol 7:19, 1990 96.
- Olsen EA, Crawford JR, Vollmer RT: Sinus histiocytosis with massive lymphadenopathy: case report and review of a multisystemic disease with cutaneous infiltrates. J Am Acad Dermatol 18:1322, 1988 97.
- Suarez CR, Zeller WP, Silberman S, et al: Sinus histiocytosis with massive lymphadenopathy: remission with chemotherapy. Am J Pediatr Hematol Oncol 3:235, 1983 98.
- Risdall RJ, McKenn RW, Nesbit ME, et al: Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 44:993, 1979 101.
- Kambouchner M, Valeyre D, Soler P, et al: Pulmonary Langerhans cell granulomatosis (histiocytosis X). Annu Rev Med 43:105, 1992 79.
- Tazi A, Bonay M, Grandsaigne M, et al: Surface phenotype of Langerhans cells and lymphocytes in granulomatous lesions from patients with pulmonary histiocytosis X. Am Rev Respir Dis 147:1531, 1993 8